Target KSVVFKAEGEHFTD-------QKGNTIVGSGSGGTT--------------
--------------------------------------------------
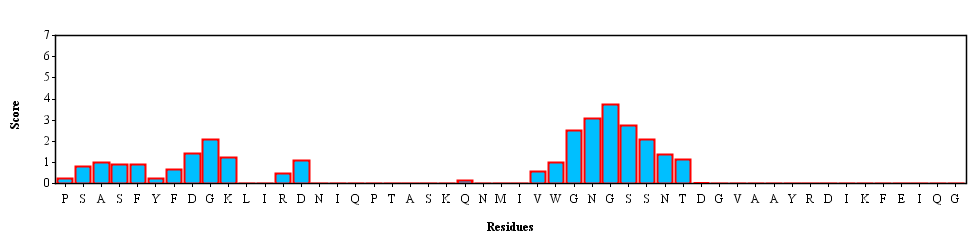
Template PSASFYFDGKLIRDNIQPTASKQNMIVWGNGSSNTDGVAAYRDIKFEIQG
Target KSVVFKAEGEHFTD-------QKGNTIVGSGSGGTT--------------
UUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUU---------------
Template PSASFYFDGKLIRDNIQPTASKQNMIVWGNGSSNTDGVAAYRDIKFEIQG
Target KSVVFKAEGEHFTD-------QKGNTIVGSGSGGTT--------------
-EEEEEETTEEEEE----EE----EEEEEE--SSS-EEEEEEEEEEEES-
Template PSASFYFDGKLIRDNIQPTASKQNMIVWGNGSSNTDGVAAYRDIKFEIQG
Target KSVVFKAEGEHFTD-------QKGNTIVGSGSGGTT--------------
--------------------------------------------------
Template PSASFYFDGKLIRDNIQPTASKQNMIVWGNGSSNTDGVAAYRDIKFEIQG
Target -KYFRIP------AMCTTSKGTIVVFADARHNTASDQSFIDT---AAARS
------------------@@@@@@@@@@@@@@---------------@@@
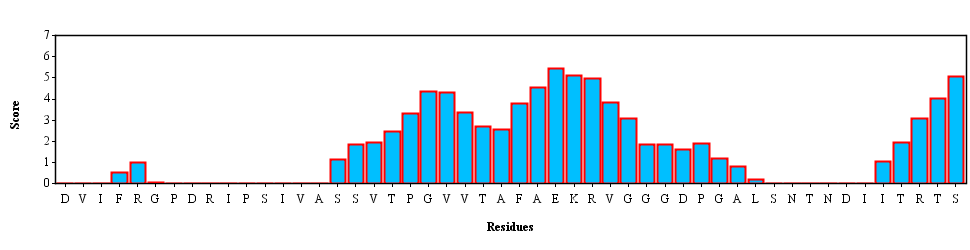
Template DVIFRGPDRIPSIVASSVTPGVVTAFAEKRVGGGDPGALSNTNDIITRTS
Target -KYFRIP------AMCTTSKGTIVVFADARHNTASDQSFIDT---AAARS
--------------------------------------------------
Template DVIFRGPDRIPSIVASSVTPGVVTAFAEKRVGGGDPGALSNTNDIITRTS
Target -KYFRIP------AMCTTSKGTIVVFADARHNTASDQSFIDT---AAARS
EEEE-TT-EEEEEEE-TTTTT-EEEEEEEEET-SSTT-TT-EEEEEEEEE
Template DVIFRGPDRIPSIVASSVTPGVVTAFAEKRVGGGDPGALSNTNDIITRTS
Target -KYFRIP------AMCTTSKGTIVVFADARHNTASDQSFIDT---AAARS
--------S--------------------S----S---------------
Template DVIFRGPDRIPSIVASSVTPGVVTAFAEKRVGGGDPGALSNTNDIITRTS
Target TDGGKTWNKKIAIYNDRVNSKLSRVMDPTCIVA
@@@@@@@@@@@@@@@@@@@----@@@@@-----
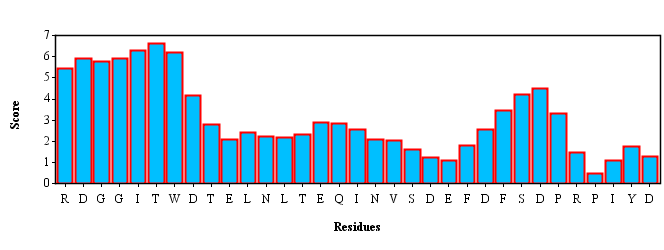
Template RDGGITWDTELNLTEQINVSDEFDFSDPRPIYD
Target TDGGKTWNKKIAIYNDRVNSKLSRVMDPTCIVA
---------------------------------
Template RDGGITWDTELNLTEQINVSDEFDFSDPRPIYD
Target TDGGKTWNKKIAIYNDRVNSKLSRVMDPTCIVA
TTSSSS----EESSTTT-SSS-EEEEEEEEEEE
Template RDGGITWDTELNLTEQINVSDEFDFSDPRPIYD
Target TDGGKTWNKKIAIYNDRVNSKLSRVMDPTCIVA
--------------------------S------
Template RDGGITWDTELNLTEQINVSDEFDFSDPRPIYD